 An online platform for predicting basic chemical properties
An online platform for predicting basic chemical properties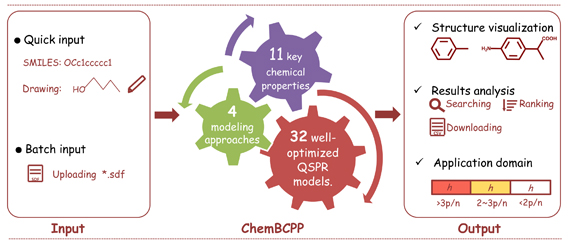
The ChemBCPP Tool (Basic Chemical Properties Prediction) has been developed to allow users to easily estimate several important chemical properties using a variety of QSAR methodologies. ChemBCPP allows a user to estimate these properties without requiring any external programs. Users can input a chemical to be evaluated by drawing it in an included chemical sketcher window, entering a structure text file, or imputing the SMILES of the chemical structures. Once a chemical has been entered, its properties can be estimated using one of several advanced QSAR methodologies. The program does not require molecular descriptors from external software packages (the required descriptors are calculated within the tool.)
Please cite: Dong J, Wang N N, Liu K Y, Zhu M F, et al. ChemBCPP: A freely available web server for calculating commonly used physicochemical properties. Chemometrics and Intelligent Laboratory Systems, 2017, 171:65-73. [PDF]
Visits since Aug 30, 2016
The recommended browsers: Safari, Firefox, Chrome,IE(Ver.>8).
E-mail: biomed@csu.edu.cn
 |
 |
 |
 |
 |
 |